本站讯 2022年1月,数学与统计学院柳军涛等在国际生物信息学重要期刊《Briefings in Bioinformatics》(IF= 11.622,中科院1区Top期刊))上发表了题为“Consensus-based clustering of single cells by reconstructing cell-to-cell dissimilarity”的论文。山东大学(威海)为论文第一完成单位,王春翔硕士,穆增超老师和牟超洲老师为论文共同第一作者,柳军涛老师为论文通讯作者。
单细胞RNA测序以单细胞的分辨率提取转录组信息,彻底改变了传统的转录组研究,它有助于识别新的细胞类型,为深入研究复杂疾病的发生发展机制、诊断和治疗提供了全新的研究思路。然而scRNA-seq数据的高维度性,以及技术层面上的生物噪声和技术噪声都会影响细胞类型的精准识别。因此,如何设计高精度聚类算法来适用不同类型的scRNA-seq数据集,同时弥补生物技术层面上的不足是目前单细胞领域亟待解决的问题。
首先,该方法基于一致性聚类(SC3),分析了不同距离测度对10个常用的单细胞基因表达数据集聚类结果的影响。结果发现,不同的距离度量方法对不同类型的基因表达数据的效果是不同的。基于此,该研究旨在为不同scRNA-seq数据集在聚类前定义适合它的最优距离度量,精准捕获细胞之间的相似性,从而实现高精度聚类。如下图所示,本文设计了SD-h算法,该算法可以通过计算d-score的分数来评估各种距离测度的可靠性,然后通过对多个距离测度进行优化综合,重构出新的综合距离矩阵。SD-h在重构的综合距离矩阵的基础上,采用层次聚类,得到了更可靠的聚类结果。
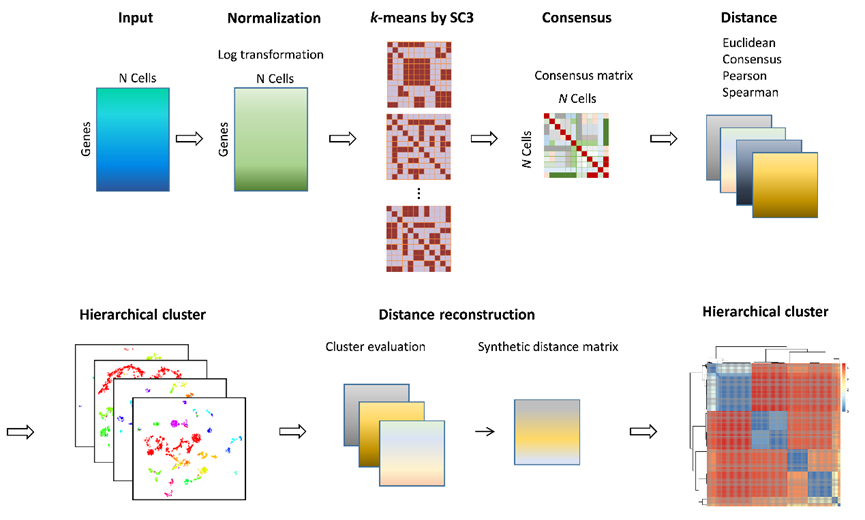
该聚类算法在9个常用的scRNA-seq数据集上进行了测试,结果表明,在大多数情况下,SD-h算法的综合距离测度比任何单一距离测度(如SC3)都能获得更准确的聚类结果,并且具有更好的普适性。例如,在Yan数据集上,SC3的ARI平均值为67.41%,而SD-h的ARI平均值为80.29%,提高幅度超过19.1%。同时,该方法还与其他六种主要聚类方法dynamictrecut、pcaReduce、tSNE+k-means、SNN-clip、SC3、Monocle和SC3的性能进行了比较。在9个数据集上的测试结果表明,SD-h在所有比较的聚类方法中,几乎在所有的数据集上表现最好,尤其在细胞数达一千以上的数据集上表现优异。
综上,该文章设计的SD-h算法通过对多个距离测度进行优化综合,重构出新的综合距离矩阵,有效地提升了聚类结果的精度,同时在多数据集上有较好的适用性。因此,该算法将在包括细胞类型识别,复杂疾病的发生发展机制、诊断和治疗得研究中起到重要的理论指导作用。
文章链接:https://academic.oup.com/bib/article-abstract/23/1/bbab379/6373558
编辑:王祎璠
